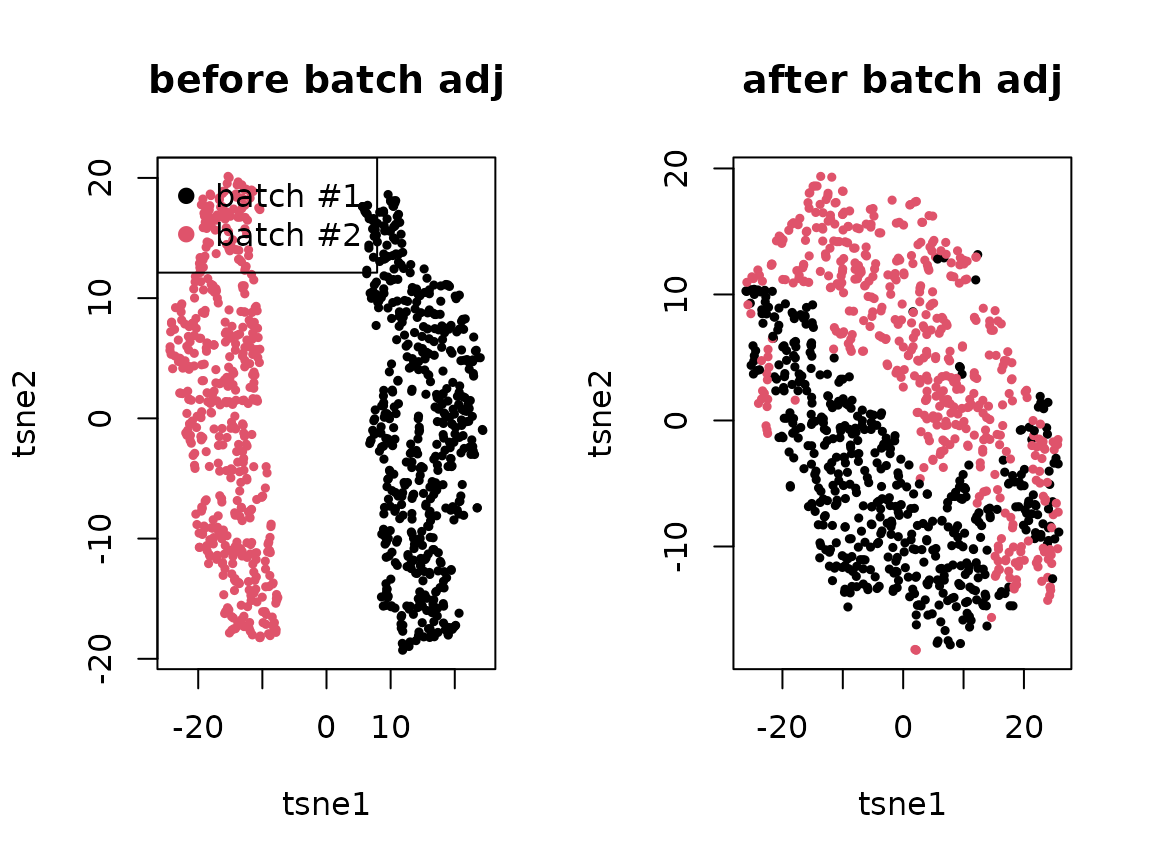2. Batch correction within each pseudobulk sample
Yongjin Park
note_batch_correction_1_concept.RmdA generative scheme for a single-cell count matrix with multiplicative batch effects
We encounter single-cell expression data consisting of multiple batches. One of the primary goals is to identify cell types (clusters/factors) and cell-type-specific gene expression patterns. However, distinguishing batch-specific and cell-type-specific genes only by a factorization method is challenging and often not identifiable from data alone. For each gene and cell , the gene expression were sampled from Poisson distribution with the rate parameter:
affected by the batch effects . More formally, letting be a batch membership matrix, assigning a cell to a batch if and only if , we assume the average gene expression rates are linearly affected by in the log-transformed space:
library(pheatmap)
set.seed(1331)
m <- 500 # genes
n <- 1000 # cells
nb <- 2 # batches
## 1. batch membership
X <- matrix(0, n, nb)
batch <- sample(nb, n, replace = TRUE)
for(b in 1:nb){
X[batch == b, b] <- 1
}
## 2. batch effects
W.true <- matrix(rnorm(m*nb), m, nb)
ln.delta <- apply(W.true %*% t(X), 2, scale)
## 3. true effects
K <- 5
.beta <- matrix(rgamma(m * K, 1), m, K)
.theta <- matrix(rgamma(n * K, 1), n, K)
lambda.true <- .beta %*% t(.theta)
kk <- apply(.theta, 1, which.max)
lambda <- lambda.true * exp(ln.delta)
yy <- apply(lambda, 2, function(l) sapply(l, rpois, n=1))
oo <- order(apply(t(.theta), 2, which.max))If we can accurately estimate a true batch effect matrix, say , it is straightforward to adjust the difference between batches. How can we identify the true batch effect for all the genes specifically expressed in the batch ? If we match cells and sampled from the batches and , respectively, we expect the batch-specific difference will persist and even amplify, but the difference originated from cell types will vanish. This problem is equivalent to estimating the potential outcome of gene expressions in each batch , .
A causal inference approach to identify batch effects
To dissect batch-specific effect in a causal inference (potential outcome) framework, we assume our confounding variables are well-distributed across different batches:
- Overlap: for all .
Moreover, we assume these covariates are sufficient enough to induce conditional dependence between potential (imputed) gene expression and batch assignment mechanisms:
- Strong ignorability: for all pairs.
Estimation of the batch effects by matching
Suppose we can counterfactually estimate gene expressions of a certain cell if the cell was measured in different batches other than the observed batch .
Like many other batch correction methods invented for single-cell RNA-seq analysis, we will assume reliably contain biologically-relevant cell state information while excluding the batch-specific effects to which the cell belong.
Observed log-likelihood:
Counterfactual log-likelihood:
Local update: Maximize batch -specific parameters
Letting ,
If and , meaning that this sample is just sampled from the batch only, and . Therefore, .
Algorithm
Initialize batch effect for each gene and batch
Initialize for each sample
Static global stat:
-
For each pseudo-bulk sample with cells ,
, ,
after matching and imputation
Iterative-updated global stat:
-
(Local step) For each PB sample :
For each :
-
(Global step) For each batch :
Repeat the previous three steps (5-7) until convergence
A toy example
Before we adjust batch membership in the random projection matrix:
[,1] [,2][1,] 0.7617260 -0.7617260 [2,] 0.8283630 -0.8283630 [3,] 0.8099248 -0.8099248 [4,] -0.7250199 0.7250199 [5,] 0.6651915 -0.6651915
## 2. regress out
##
## X theta = X inv(X'X) X' Y
## = U D V' V inv(D^2) V' (U D V')' Y
## = U inv(D) V' V D U' Y
## = U U' Y
x.svd <- svd(X)
U <- x.svd$u
U.t <- t(x.svd$u)
Q.t <- t(Q.raw)
Q.t <- Q.t - U %*% U.t %*% Q.t
Q <- t(Q.t)After we adjust the batch effects:
cor(Q.t, X) [,1] [,2][1,] -2.276319e-16 2.276319e-16 [2,] -5.524025e-16 5.524025e-16 [3,] -2.934286e-16 2.934286e-16 [4,] 3.075415e-16 -3.075415e-16 [5,] -3.636223e-16 3.636223e-16
q.svd <- svd(Q)
## 3. sorting
B <- (sign(q.svd$v) + 1)/2
ss <- apply(sweep(B, 2, 2^(seq(0,K-1)), `*`), 1, sum) + 1
feat.dn <- apply(Q, 2, function(x) x / sqrt(sum(x^2)))
knn <- 3
d <- nrow(feat.dn)
library(RcppAnnoy)
## a. construct dictionary for each batch
dict.list <- lapply(sort(unique(batch)),
function(b) { new(AnnoyAngular, d) })
for(j in 1:length(batch)){
b <- batch[j]
dict.list[[b]]$addItem(j, feat.dn[,j])
}
for(dd in dict.list){
dd$build(50)
}
## b. a simplified routine to retrieve and estimate counterfactual y
.counterfactual <- function(j){
v <- feat.dn[,j]
nn <- c()
dd <- c()
for(k in 1:nb){
if(k == batch[j]) next
.nn <- dict.list[[k]]$getNNsByVector(v, knn)
.dd <- apply(feat.dn[, .nn], 2, function(u) sum((u - v)^2))
nn <- c(nn, .nn)
dd <- c(dd, .dd)
}
w <- exp(-(dd - max(dd)))
w <- w/sum(w)
yy[, nn, drop = FALSE] %*% matrix(w, ncol=1)
}
ngene <- nrow(yy)
nbatch <- ncol(X)
nsample <- max(ss)
.delta.db <- matrix(1, ngene, nbatch) # gene x batch effects
.delta.num.db <- matrix(0, ngene, nbatch) # gene x batch numerators
.delta.denom.db <- matrix(0, ngene, nbatch) # gene x batch denominators
.prob.bs <- matrix(0, nbatch, nsample) # batch x sample probabilities
.size.bs <- matrix(0, nbatch, nsample) # batch x sample freq
.ybar.ds <- matrix(0, ngene, nsample) # gene x sample observed average
.zbar.ds <- matrix(0, ngene, nsample) # gene x sample imputed average
.mu.ds <- matrix(1, ngene, nsample) # gene x sample adjusted average
## Precalculate some statistics
for(s in 1:nsample){
if(sum(ss == s) < 1) next
.yy <- yy[, ss == s, drop = FALSE]
.zz <- do.call(cbind, lapply(which(ss == s), .counterfactual))
.ybar.ds[,s] <- apply(.yy, 1, mean)
.zbar.ds[,s] <- apply(.zz, 1, mean)
.prob.bs[,s] <- colMeans(X[ss == s, ])
.size.bs[,s] <- colSums(X[ss == s, ])
.y.dsb <- yy[, ss == s, drop = FALSE] %*% X[ss == s, , drop = FALSE]
.delta.num.db <- .delta.num.db + .y.dsb
}
.gamma.ds <- matrix(1, ngene, nsample)
for(iter in 1:100){
.mu.ds <- (.ybar.ds + .zbar.ds) / (.delta.db %*% .prob.bs + .gamma.ds + 1e-8)
.gamma.ds <- .zbar.ds / (.mu.ds + 1e-8)
.delta.db <- .delta.num.db / (.mu.ds %*% t(.size.bs) + 1e-8)
}Can we recover the original batch effects?
par(mfrow=c(1,2))
plot(.delta.db[,1], W.true[,1], pch=19, xlab="estimated delta", ylab="true delta effect", main="batch1")
plot(.delta.db[,2], W.true[,2], pch=19, xlab="estimated delta", ylab="true delta effect", main="batch2")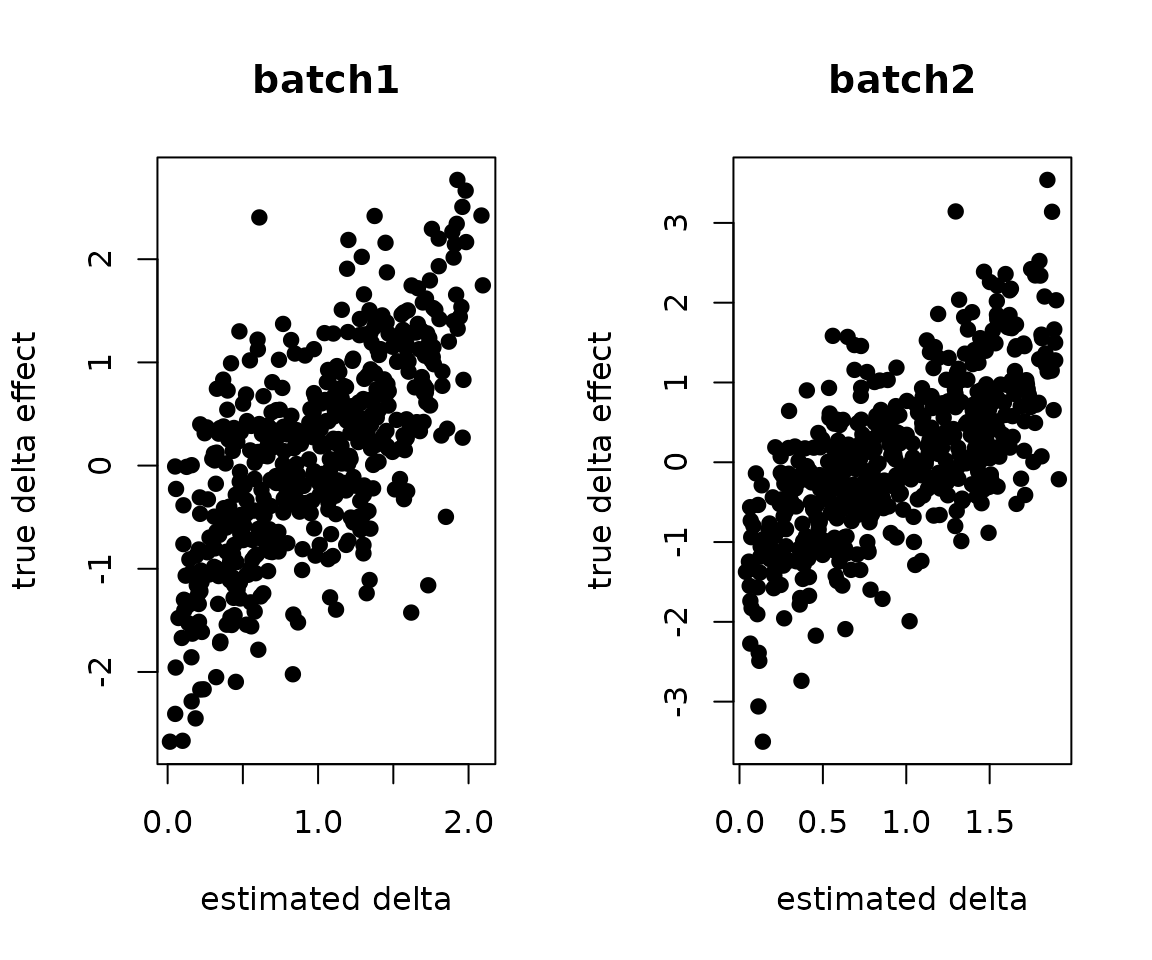
Are they independent of the cell type effects?
y.true <- sweep(lambda.true %*% X, 2, colSums(X), `/`)
par(mfrow=c(1,2))
plot(.delta.db[,1], y.true[,1], pch=19, xlab="estimated delta", ylab="true y mean", main="batch1")
plot(.delta.db[,2], y.true[,2], pch=19, xlab="estimated delta", ylab="true y mean", main="batch2")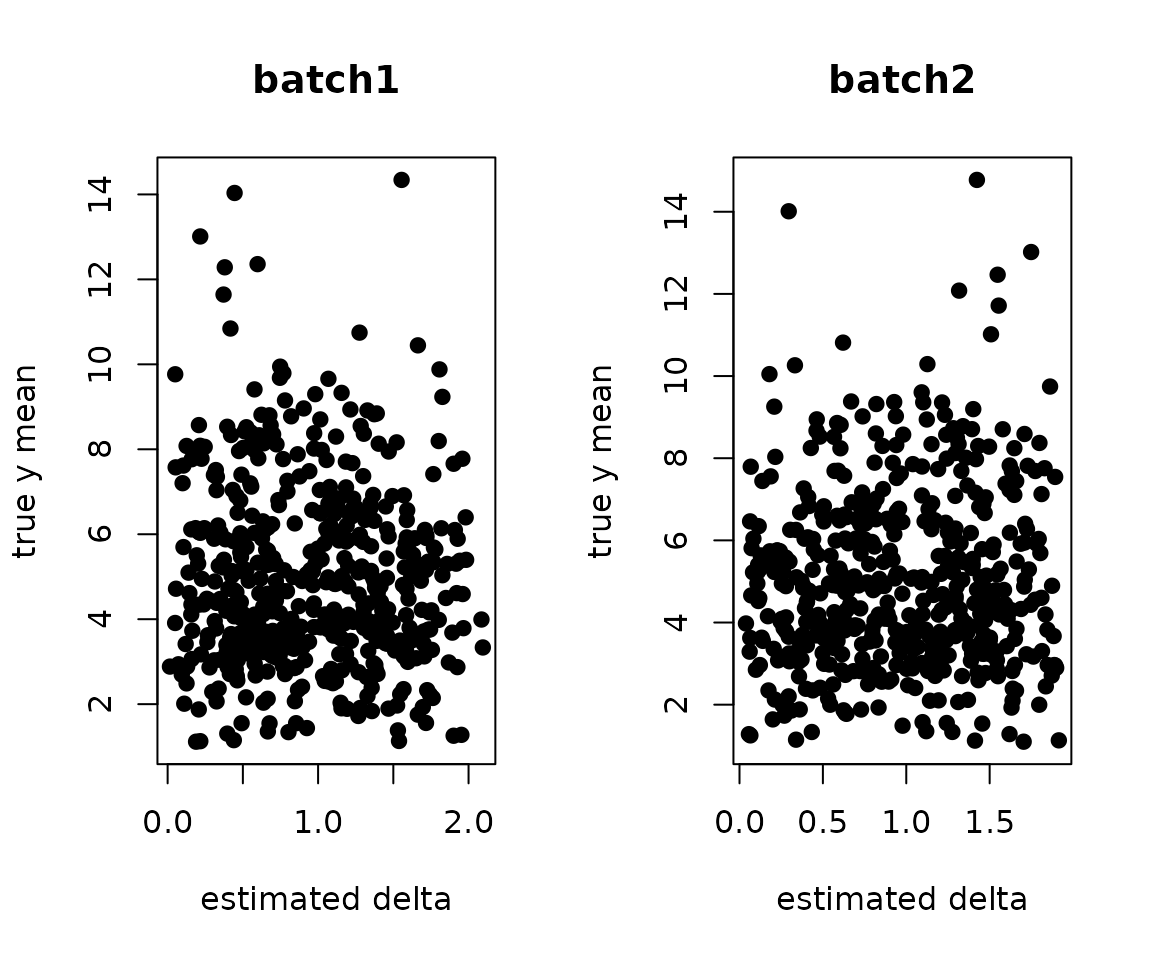
While adjusting the estimated batch effects, can we recover the unbiased cell type effects? The following is before adjustment:
ybar <- sweep(yy %*% X, 2, colSums(X), `/`)
par(mfrow=c(1,2))
plot(ybar[,1], y.true[,1], pch=19, xlab="sample mean", ylab="true y mean", log="x", main="batch1")
plot(ybar[,2], y.true[,2], pch=19, xlab="sample mean", ylab="true y mean", log="x", main="batch2")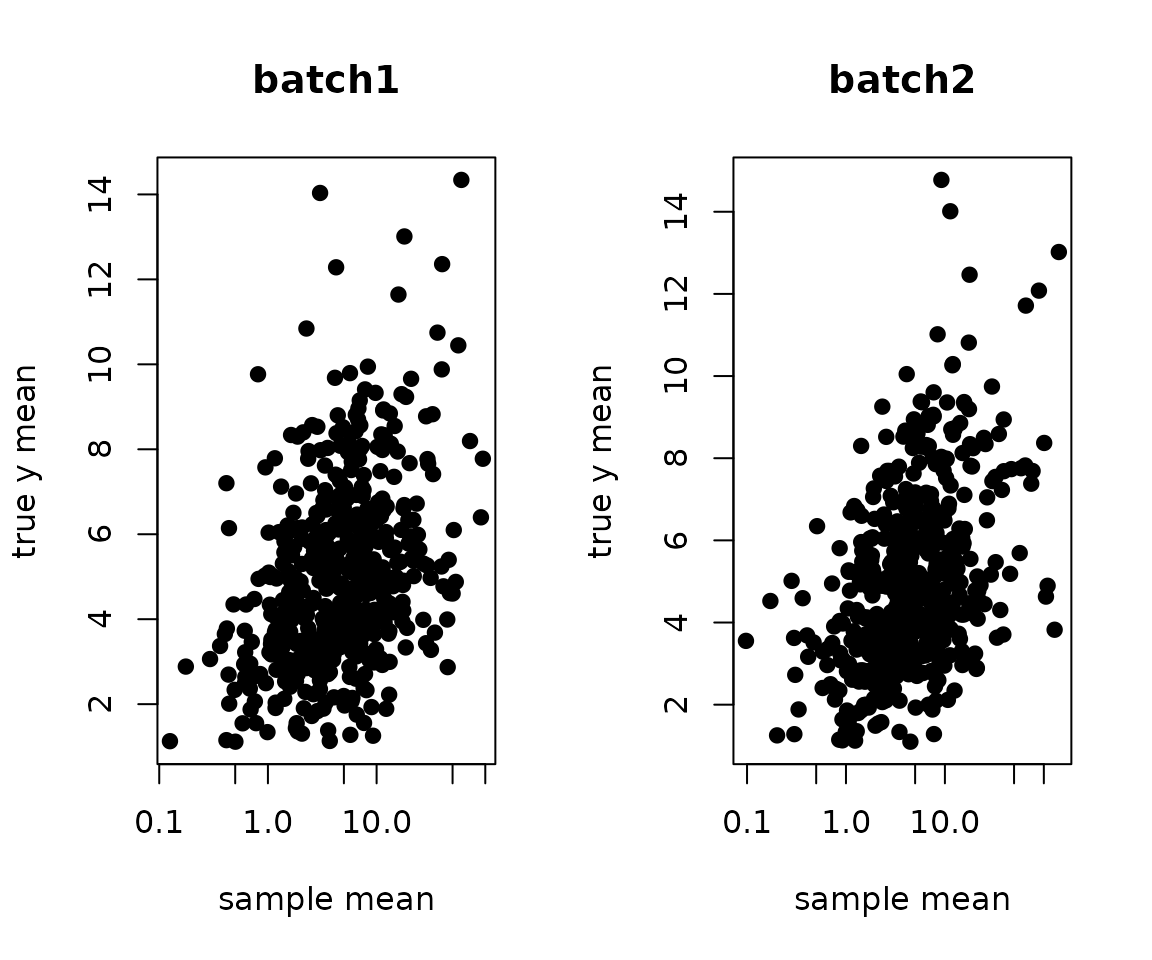
Here, we adjusted the batch effects:
ybar.adj <- sweep((yy / .delta.db[, batch]) %*% X, 2, colSums(X), `/`)
par(mfrow=c(1,2))
plot(ybar.adj[,1], y.true[,1], pch=19, xlab="adjusted sample mean", ylab="true y mean", log="x", main="batch1")
plot(ybar.adj[,2], y.true[,2], pch=19, xlab="adjusted sample mean", ylab="true y mean", log="x", main="batch2")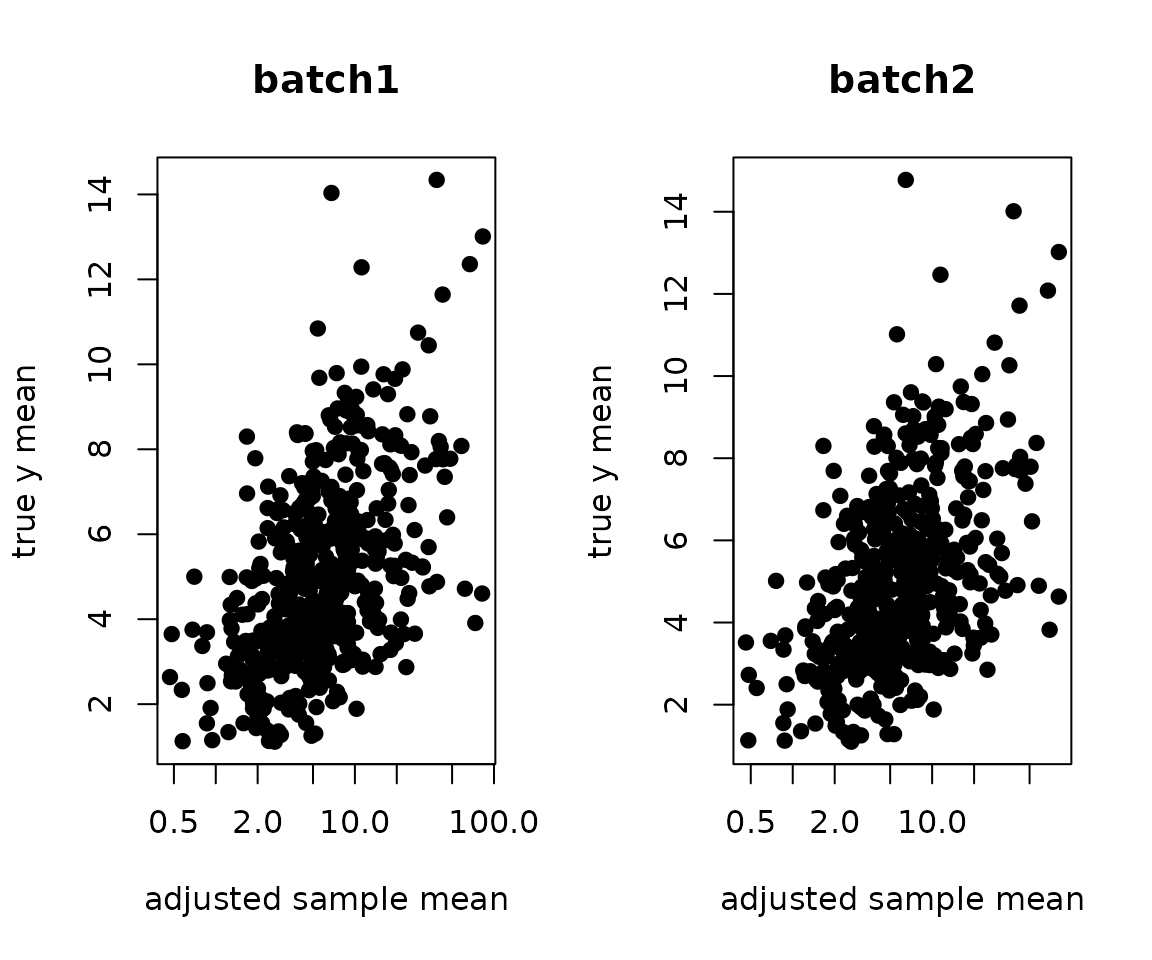
par(mfrow=c(1,2))
.tsne <- Rtsne::Rtsne(log(1 + t(yy)), num_threads=4)$Y
plot(.tsne[,1], .tsne[,2], col=batch, pch=19, cex=.5, main="before batch adj",
xlab = "tsne1", ylab = "tsne2")
legend("topleft", c("batch #1", "batch #2"), col=1:2, pch=19)
.tsne <- Rtsne::Rtsne(log(1 + t(yy/.delta.db[,batch])), num_threads=4)$Y
plot(.tsne[,1], .tsne[,2], col=batch, pch=19, cex=.5, main="after batch adj",
xlab = "tsne1", ylab = "tsne2")